Basic Biologic Research
Without a basic understanding of biology the possibilities for innovation that can change our lives and health become limited. We strive in each of our projects to open new spaces in basic biologic research that address fundamental questions about mechanisms in biology. We are developing new biological approaches and biotechnologies to open unexplored and understudied biological mechanisms.
RNA-binding by arginine/glycine-rich (RGG/RG) domains recent surveys have revealed that RGG/RG domains may be the second most common RNA-binding domain found in nature. This domain has hitherto escaped rigorous investigation. Our RNA-binding proteins FUS, EWSR1, and TAF15 each contain three highly conserved RGG/RG domains, making them the ideal prototypic proteins for this research aim. We have characterized the mechanism for binding to RNA by domains in FUS, FMRP, and hnRNPU. We find RGG/RG domains to consistently prefer binding to complex RNA structures containing both double stranded regions and nearby perturbations to the structure – e.g. junctions, loops, bulges, and in non-biological targets, g-quadruplexes. Among these broadly defined specificities, these highly flexible domains display little of any additional preference; thus, we define the RGG/RG domain as possessing degenerate specificity. Our ongoing projects include: (1) define how highly conserved RGG/RG domains with specific proteins contribute to a specificity across broad subsets of the transcriptome, and (2) determine the mechanism by which RNA recognition by RGG/RG domains control the ultimate function of the proteins.

RGG/RG domains are common in RNA-binding domains: RNA-recognition motifs (RRM) is the most abundant RNA-binding domain (RBD) and frequently found in the heterogeneous ribonucleoprotein particle (hnRNP) family of proteins (Left). RGG/RG domains also frequently appear in proteins possessing other RBDs and may broaden the specificity of these proteins. (Right) The RGG/RG domains of FET proteins (FUS, EWSR1, and TAF15) are conserved beyond vertebrates.
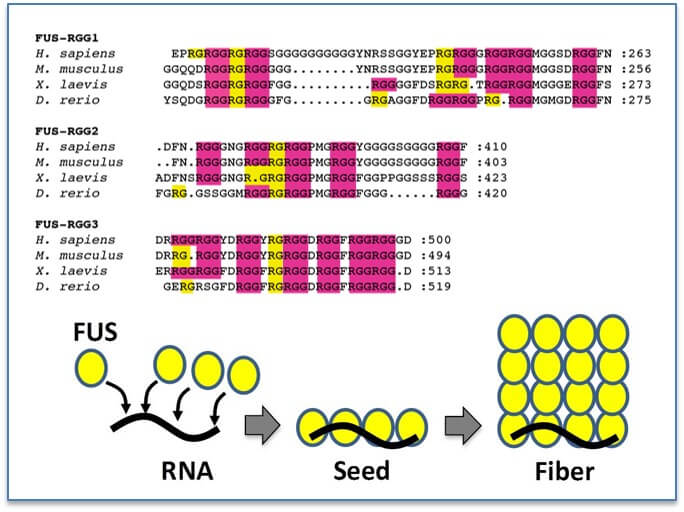
RGG/RG domains are conserved: RGG/RG domains are enriched in arginines and glycines and yet show diversity in the number and spacing of each RGG/RG. However, for specific proteins this sequence remains conserved suggesting the number and positioning of RGG/RG repeats is not random and has functional significance (Akdogan et al., in submission). Binding of RNA by the RGG/RG domains induces a structural shift in FUS that seeds oligomerization.
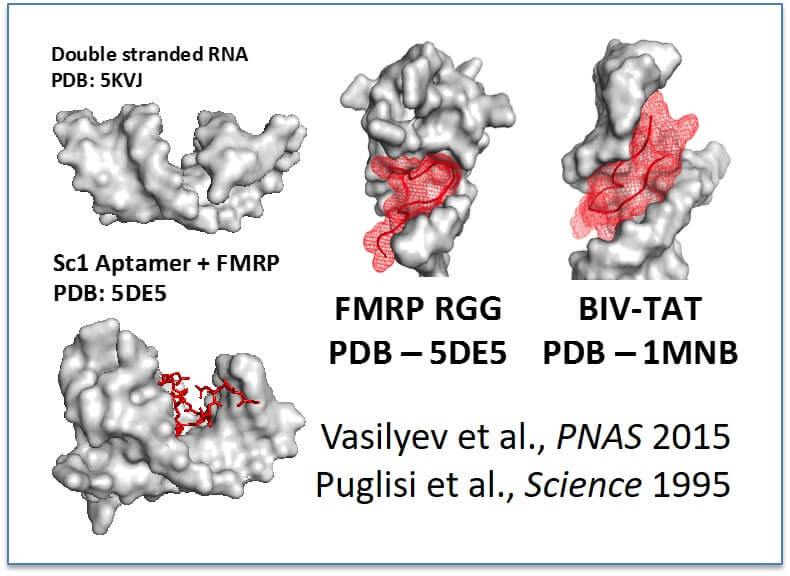
Working model for RGG/RG recognition of RNA: RGG/RG domains bind complex RNA structures containing double stranded RNA and do not bind simple double stranded RNA (Akdogan et al., in submission). We hypothesize that two published structures of RGG/RG and RNA complexes demonstrate a general model that nearby structures serve to widen the major grove allowing access for RGG/RG domains (reference shown, left).
Relevant Publications:
Akdogan B, Thompson VF, Ahmed N, White C, Batey RT, Schwartz JC, “Intrinsically disordered RGG/RG domains mediate degenerate specificity in RNA binding”, under review
Wang X, Schwartz JC, Cech TR, “Nucleic acid-binding specificity of human FUS protein.” NAR 2015; 43(15):7535-43. (PMID: 26150427)
Schwartz JC, Cech TR, Parker RR, “Biochemical properties and biological functions of FET proteins”, Annu. Rev. Biochem. 2015; 84:355-79. (PMID: 25494299)
Schwartz JC, Wang X, Podell ER, Cech TR, “RNA seeds higher order assembly of FUS protein” Cell Reports, 2013; 5(4):918-25. (PMID: 24268778)
A fundamental property of life on Earth and a defining characteristic for eukaryotic cells is sequestering their biochemical functions using membranes comprised of lipids. However, also found throughout life are organelles that sequester their biological functions without the benefit of lipid membranes. These are non-membrane bound organelles (a.k.a granules), and examples include nucleoli, cajal bodies, nuclear speckles, stress granules, and p-bodies. Our work has revealed the mechanisms by which proteins containing a novel domain, dubbed a low complexity (LC) domain, spontaneously form these granular bodies in vitro. These in vitro assembled granules we refer to as droplets. This discovery offers the unprecedented opportunity to study the biophysical properties of cellular granules and elucidate answers to fundamental questions concerning their function. The biophysical properties of cellular granules have remained baffling as they maintain their integrity while also displaying remarkable liquid-like behaviors. Our ongoing and future planned projects are to (1) characterize the biophysical properties driving droplet formation, and by extension – granular bodies, (2) isolate novel granular bodies comprised of proteins possessing functions unique from previously known cellular granules, such as transcription granules serving as a molecular scaffold for the recruitment of the transcription machinery, and (3) seek out and survey novel droplet forming proteins comprising novel granular bodies in human, invertebrate, and prokaryotic organisms.
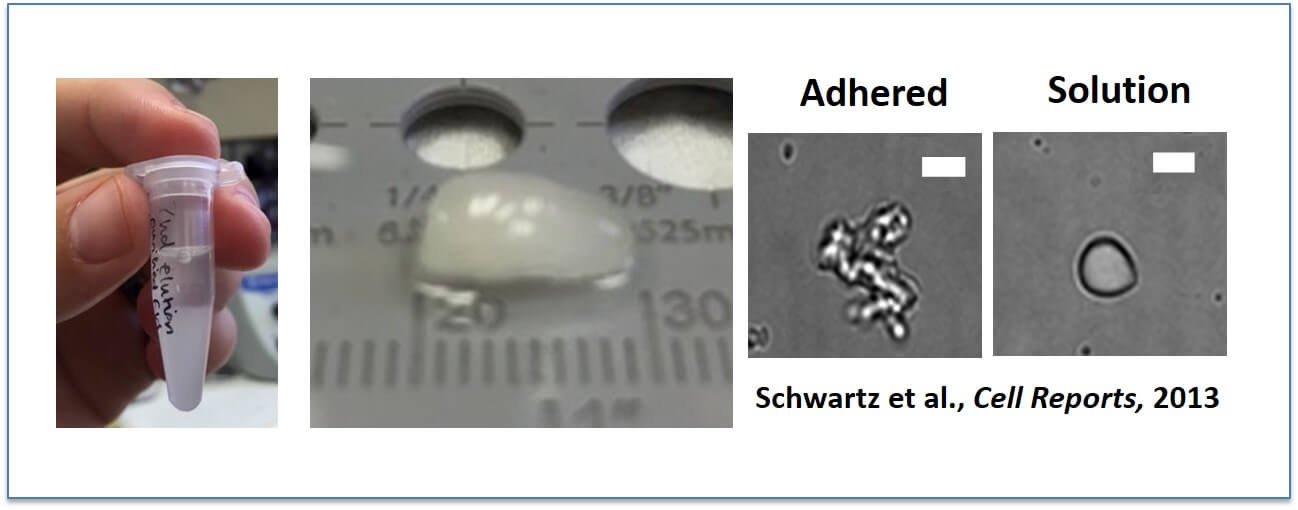
FUS spontaneously assembles into liquid-like droplets and then hydrogels: Yes, that is protein. FUS spontaneously assembles into these (Left above), particularly at low temperatures (near freezing). To study these assemblies in vitro, we prefer to allow them to adhere to glass surfaces in order that we can perform binding assays, in which case they have an amorphous shape (second from right). But when unattached from the glass, they can “snap” back into spherical shapes and hence earned the name “droplets”.
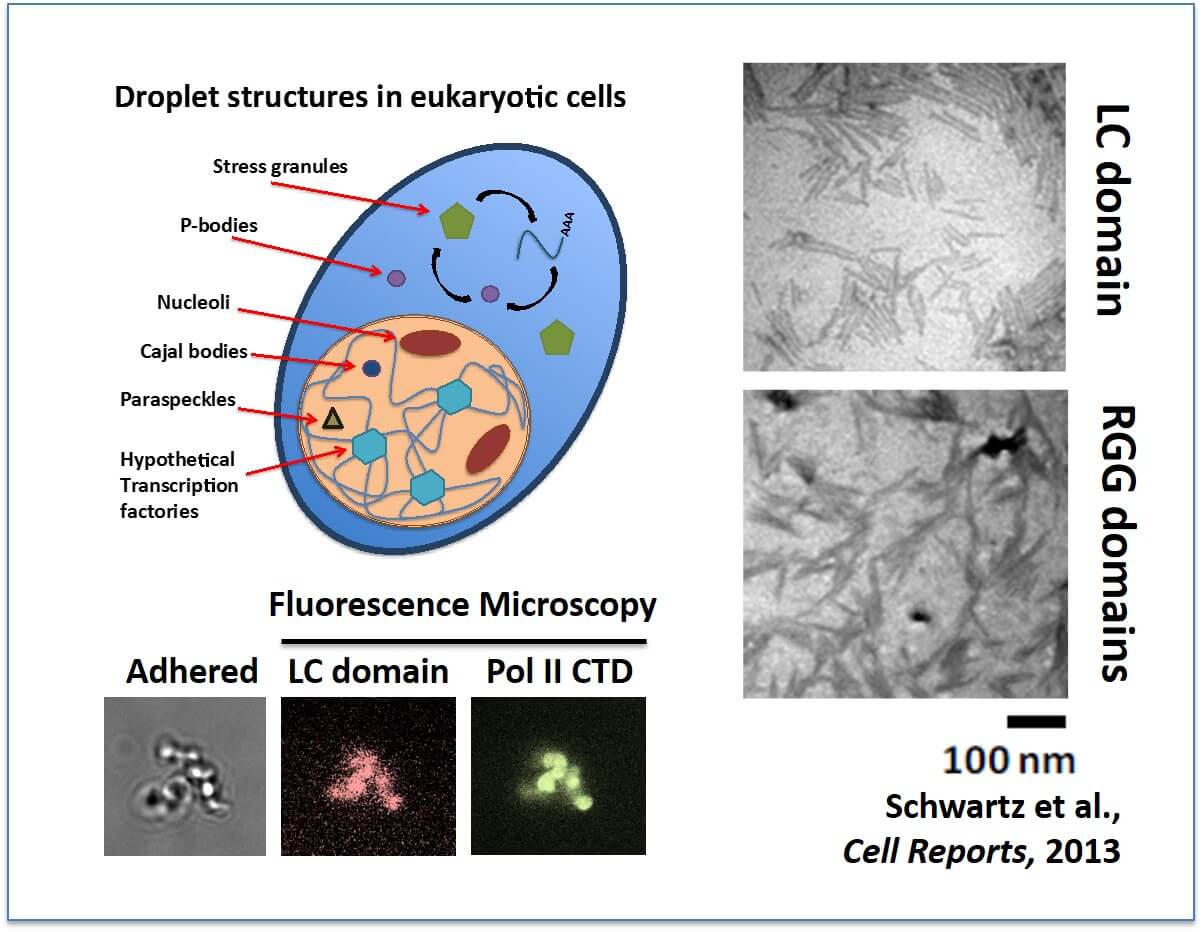
Many non-membrane bound organelles compartmentalize cellular biology: (Left) Numerous granular bodies have been described in cells, particularly within the nucleus. One function of FUS in cells is to bind the C-terminal domain (CTD) of RNA Pol II to effect transcription. We can demonstrate this binding using our in vitro assembled droplets. Thus, we hypothesize a novel cellular granule that assembles around active gene promoters. (Right) The LC domain of FUS assembles into protein fibers. RGG/RG domains assemble into fibers as well. Both bind the CTD of RNA Pol II, the significance of which is currently unknown.
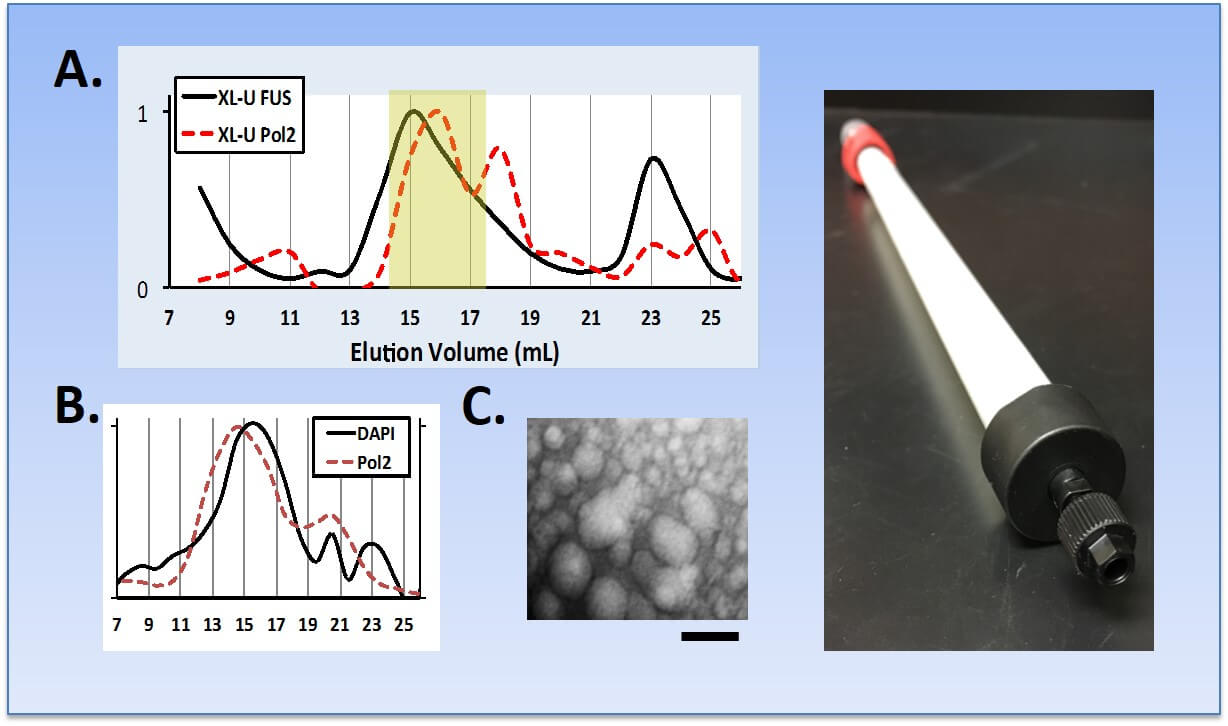
Purification of FUS droplets from cells. (A) Cross-linked cell lysates were fractionated by SEC with FUS and RNA Pol II eluting together in complexes between 70-150 nm complexes. For comparison, the crystal structure of the mammalian RNA Pol II holoenzyme is only 25 nm in diameter. (B) Small DNA fragments are protected from degradation by benzonase or sonication and co-eluted with RNA Pol II complexes. Consistent with this observation, next-generation sequencing reveals that these fragments are enriched for genomic regions that are also enriched for RNA Pol II. (C) Droplets were visualized by TEM and diameters confirmed (bar = 100 nm). (Right) In violation of all that is holy in SEC chromatography, we have developed a custom SEC protocol for separating and detecting very large complexes of RNA Pol II and other granular bodies in nuclear and whole cell lysates.
Relevant Publications: (manuscript in preparation)
Schwartz JC, Cech TR, Parker RR, “Biochemical properties and biological functions of FET proteins”, Annu. Rev. Biochem. 2015; 84:355-79. (PMID: 25494299)
Schwartz JC, Wang X, Podell ER, Cech TR, “RNA seeds higher order assembly of FUS protein” Cell Reports, 2013; 5(4):918-25. (PMID: 24268778)
AMYOTROPHIC LATERAL SCLEROSIS (ALS)
To say ALS is a “devastating” neurodegenerative disease hardly covers it. It is relentless. There is no cure. There’s no therapy that can slow the progression of this disease. There’s not even bioassay to definitively diagnose the disease. More than 5000 patients are diagnosed with ALS in America each year. The average patient lives for three years following diagnosis. The death of motor neurons lead to continued paralysis and loss of muscle control, known as muscle atrophy. Diagnosis can take more than 6 months, largely due to our poor understanding of what mechanisms underlie this disease. This lost time is both expensive and prevents early, life-extending care that would have offered patients a higher quality of life and extend life up to two years beyond the standard prognosis.

Thanks to modern advances in genome-wide association studies (GWAS), the ALS community has recently been provided a list of proteins for which mutations cause ALS – most often for the minor fraction of families inherit these genes through families (familial ALS), but also for “sporadic” cases, where a patient is born with a spontaneous, disease-causing mutation not found in his/her parent’s genomes. These mutations give us the ability to recreate ALS pathology in a controlled environment – either in human cells grown in the lab or in mice. However, the majority of the proteins now known to play a role in ALS have not previously undergone rigorous study related to their normal function. This limitation prevents the development of hypotheses concerning the mechanism for the mutant proteins to contribute to disease. RNA-binding proteins comprise the largest fraction of those implicated in ALS, and the study of RNA-binding proteins is the core specialty of the Schwartz lab.
Our lab engages in “holistic” research, spanning the test tube to human cells, patient samples, and animal models. The research from the test tube provides us a precise mechanism to explain phenotypes observed in cell and physiological models. The animal models in turn provide limits, directing whether a specific mechanism is a significant contributor to phenotypes consistent with ALS pathology. In this, we aim to be as relentless as ALS in the fight to stop this disease.

Amyotrophic Lateral Sclerosis (ALS): ALS is a disease of neurons that control muscle movement, called motor neurons. Motor neurons reside both in the cortex and in the spinal cord. Loss of muscle control and muscle wasting (atrophy) is due to motor neurons dying. The disease progresses rapidly with most patients surviving less than 3 years after diagnosis. Age of onset varies considerably. Within Tucson AZ, approximately 15 cases of ALS diagnosed each year. At any time, approximately 25 to 30 patients are currently living with ALS (KS personal communication).
The first protein we’ve characterized related to ALS is FUS. Discovered to be involved in ALS in 2009, mutations in FUS remain tied as the third leading cause of familial ALS. Our earliest studies found FUS to be a broad regulator of transcription in cells. We then found that RNA stimulates FUS to form fibrous assemblies and this activity is required for FUS interactions with the transcription machinery. Armed with this knowledge, we explored cells recovered from skin biopsies donated by ALS patients to research. We found that FUS assemblies are altered in ALS patients, losing their dynamics, their ability to assemble and disassemble rapidly, and they no longer function effectively as regulators of transcription. Ongoing projects address: (1) the role of FUS in transcription, RNA processing, and ALS pathology; and (2) development of approaches to purify FUS containing granules out of cells, both non-disease and cells derived from ALS patients.

The Normal Function of FUS: (A) FUS is a multi-domain RNA-binding protein with an N-terminal Low Complexity (LC) domain. The most prevalent ALS-causing mutations in FUS are found in the nuclear localization signal (NLS). (B) FUS binds RNA Pol II (RNAP2) and co-localizes with the polymerase to active gene promoters. (C) FUS is one of the most abundant proteins found in the nucleus. It targets transcription by recognizing the earliest transcripts produced by RNA Pol II, and then forms a fibrous scaffold, which recruits more RNA Pol II, other transcription machinery, and regulates phosphorylation of the CTD of RNA Pol II.

Study of FUS function in ALS patient-derived cells: (A) An invaluable resource to ALS research are fibroblast cells donated by ALS patients. (B) Despite mutations to the nuclear localization signal of FUS, in patient samples most of the FUS protein remains in the nucleus. Whereas pathology had been presumed to be associated with toxic cytoplasmic activity of the mislocalized protein, we have explored whether nuclear dysfunction may contribute significantly to pathology.

Aberrant FUS function in ALS patients: Despite the majority of FUS being in the nucleus in ALS patients bearing FUS mutations (A), nuclear FUS is trapped in aggregates that cannot migrate on an SDS-PAGE gel and not functional (B). (C) Genes actively transcribed by RNA Pol II are localized within the cell nucleus to discrete foci and this open chromatin stains weaker for DAPI. For ALS patients with FUS mutations, this organization is disrupted and the foci of RNA Pol II becomes fragmented. (D) Our model is that FUS forms droplets, which are scaffolds surrounding expressed genes. We find in ALS patient cells, FUS droplets in the nucleus become aggregate and no longer function to regulate transcription or help organize the transcription machinery.
Relevant Publications:
Schwartz JC, Cech TR, Parker RR, “Biochemical properties and biological functions of FET proteins”, Annu. Rev. Biochem. 2015; 84:355-79. (PMID: 25494299)
Schwartz JC, Podell ER, Han SSW, Berry JD, Eggan KC, Cech TR, “FUS is sequestered in nuclear aggregates in ALS patient fibroblasts” Mol Biol Cell, 2014; 25(17):2571-8. (PMID: 25009283)
Schwartz JC, Wang X, Podell ER, Cech TR, “RNA seeds higher order assembly of FUS protein” Cell Reports, 2013; 5(4):918-25. (PMID: 24268778)
Schwartz JC, Ebmeier CC, Podell ER, Heimiller J, Taatjes DJ, Cech TR, “FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2” Genes Dev, 2012; 26:2690-95. (PMID: 23249733)
Tied with FUS as the third leading cause of familial ALS is the RNA-binding protein TDP-43. We hypothesize that it is the intersection of overlapping functions for proteins involved in ALS pathology that will reveal to us the molecular basis for this disease. Moreover, TDP-43 aggregation is a nearly ubiquitous feature found in ALS patients, even those who do not possess the disease-causing mutations in this gene. Finally, TDP-43 provides the link connecting ALS pathology to other neurodegenerative diseases and notably, dementia. It is now appreciated that Frontal Temporal Dementia (FTD) is a disease that is closely related to ALS. Some mutations in the same genes, such as TDP-43, cause both diseases. Pathological aggregation of the same proteins, such as FUS, is observed in both diseases. One shared function between FUS and TDP-43 is the ability to regulate gene expression at the level of transcription. We are exploring whether TDP-43 possess other functional overlaps with FUS or pathological mechanisms, including those involving DNA damage repair. Our hypothesis is that while each ALS-associated gene has many cellular functions, it is the overlap of those where the most likely mechanism of disease will lie.

Affinity-enrichment mass spectrometry reveals interactions shared by FUS and TDP-43: Use of improved mass spectrometry approaches greatly enhances sensitivity. Our preliminary analysis of recent data revealed known interactors, such as NONO, FXR1, and hnRNPU. We also found interactors with known significance for ALS pathology, such as hnRNPA2B1, PPIA, GABBR1, and TUBBA4A.

The role of TDP43 in transcription regulation: (A) Using a next-generation sequencing technique, GRO-seq, we can visualize transcription as it is occurring across the entire genome. Then by removing TDP-43 by siRNA knockdown, we can determine the affect TDP43 has on transcription. (B) Multiple models may explain TDP43 regulation of transcription. TDP43 may be recruited to regulate transcription by binding the transcribed RNA (i) or the DNA (ii). It may bind enhancer RNAs (eRNA) upstream of the gene (iii). TDP43 is a constitutive member of paraspeckles, which raises the possibility of a connect between these bodies and transcription (iv, West et al., J Cell Biol 2016). (C) By replacing TDP43 with those deleting key domains, the mechanism of transcription regulation can be further elucidated.
A pressing question about ALS pathology is why do only neurons die and why does the disease mostly appear in old age. The hypothesis that DNA damage repair defects might contribute to the pathology neatly connects these two observations from the clinic. Recently, studies from many labs support the conclusion that DNA damage in the nervous system accumulates with age and the mechanisms for DNA damage repair seem to weaken with age. In fact, several of the genes implicated in ALS have links to DNA damage repair. The most well known connection is that FUS is required for DNA damage repair and is one of the earliest proteins recruited to a DNA damage site. A recent paper has suggested and we have multiple lines of evidence supporting a role for TDP-43 in DNA damage repair (Hill et al., PNAS 2016).

Multiple genes implicated in ALS are known to be involved in ALS pathology: FUS, VCP, and SETX have all been implicated as directly interacting with DNA repair machinery. SOD1 and p62 have roles in autophagy, which is linked to DNA damage through regulating the production of radical oxidative species (ROS). The role of TDP-43 remains to be determined.

Identification of proteins interacting with FUS and TDP-43 after DNA damage: Heat maps of z-scores indicate change in levels of proteins detected after DNA damage, with green increases and red decreases. Proteins where changes rose to the level of statistic significance indicated by blue bars. Currently, we hypothesize that FUS and TDP-43 cohabit several nuclear bodies, though not interacting directly. After DNA damage, FUS leaves many of its unique complexes and increases its association with complexes that also contain TDP-43. Few interactions for TDP-43 significantly change. Of known interactors of FUS and TDP-43, paraspeckles and the snRNP complex seem unchanged by DNA damage, RNA exosome interactions increase, and hnRNP interactions decrease.
EWING'S SARCOMA
That a link may exist between two completely unrelated pathologies, ALS and Ewing sarcoma, is a case of truth being stranger than fiction. Ewing's sarcoma is the second most common pediatric bone cancer and diagnosed in more than 200 patients each year, mostly children. The cause of Ewings sarcoma is an oncogenic fusion event involving either FUS or its close homologue, EWSR1.
This fusion event replaces the RNA-binding domains of EWSR1 or FUS with a DNA-binding domain of a developmental transcription factor, Fli1, to make EWS-Fli1 or FUS-Fli1. Ewing’s sarcoma is characterized as having a “quiet genome”, with few additional mutations being found other than this fusion event. This makes Ewing’s sarcoma one of the most minimal biochemical models of sarcoma pathology. Our work explores how the normal function of EWSR1 and FUS is short-circuited and redirected towards the genetic targets of the Fli1 protein, producing tumor development that rapidly develops into malignancy and metastasis.
EWS-Fli1 and FUS-Fli1 retain the abilities of their original proteins to polymerize and form droplets. These proteins are also transcription regulators, both activating and repressing gene expression in cells. Like EWSR1 and FUS, we are exploring whether the active form of the Fli1 fusion proteins that affect transcription are the polymers the proteins, and whether these proteins interact with the wild-type FET proteins (FUS, EWSR1, and TAF15) to assist in their function. Because Fli1 fusion proteins have the ability to both activate and repress transcription, we are exploring whether recruitment of alternate transcription factors mediate the switch in outcomes. Moreover, we are testing the ability of the Low Complexity (LC) domain of FUS or EWSR1 to stimulate chromatin remodeling in cells and how that contributes to reprogramming transcription in these cells towards tumorigenesis.

Formation of protein polymers by FUS-Fli1 and EWS-Fli1: (A) The translocation event driving Ewing’s sarcoma fuses the LC domain of FUS or EWSR1 to the DNA-binding domain (DBD) of Fli1, an ETS family transcription factor. (B) In Ewing’s sarcoma, Fli1 fusion proteins bind repetitive GGAA DNA sequences, which raises the hypothesis that these sequences promote oligomerization of the FET-fusion proteins along the DNA. Therefore, like FUS and EWSR1, it may be that the oligomers of FUS-Fli1 and EWS-Fli1 are the active forms of the proteins. Recruitment of alternative co-factors may explain the difference in outcomes to either activate or repress gene expression. (C) We have formed in vitro reconstituted droplets of FET proteins or their LC domains. The LC domain readily incorporates into FUS droplets, suggesting that Fli1 fusion proteins may seed higher order interactions with other FET proteins, including FUS and EWSR1.

FET proteins, and their oncogenic fusions, form phase-separated droplets: The oligomer form of FET proteins are active form to regulate transcription (A). Oligomers of FET proteins coalesce into phase-separated droplets that serve as a molecular scaffold and concentrates binding partners in vitro and in cells (B). The underlying organization of proteins within these droplets remains to be determined. (C) In Ewing’s sarcoma, we hypothesize that the Fli1 fusion proteins reorganize chromatin to form aberrant regions of active gene expression, thereby reprograming mesenchymal stem cells into Ewing’s sarcoma.
Surprisingly, not only are the genetic causes of Ewing’s sarcoma shared with those of ALS, both Ewing’s sarcoma and certain ALS patients show a defect in DNA damage repair. In the case of Ewing’s sarcoma, this is a mechanism that has been and continues to be a target for therapeutic approaches. Currently, induction of DNA damage through radiation treatment is the primary therapeutic strategy in treating Ewing’s sarcoma, and this produces a 65 to 70% survival rate over 5 years. This prognosis for Ewing’s sarcoma patients has not much improved in 30 years. The mechanism underlying Ewing’s sarcoma’s deficit in DNA damage repair is difficult to postulate. Recently, combinatorial treatment of Ewing’s sarcomas with DNA damage agents and inhibitors of the enzyme PARP have proven to be more effective than DNA damage alone (Stewart et al., Cell Reports 2014). We are testing whether the role of EWS-Fli1 in transcription regulation is linked to its function in DNA damage repair. We are screening a panel of small molecules targeting transcription factors to search for combinations with greater and more synergistic effects. This approach both is revealing about the mechanism by which DNA damage repair is impaired in Ewing’s sarcoma and has the potential to produce new therapeutic strategies for clinical development.

Targeting FET protein functions increase Ewing’s sarcoma sensitivity to DNA damage: (A) Our first drug hit RS003 has a synergistic effect with the DNA damage agent SN38 inducing potent cell death at 96 hours for Ewing’s sarcoma but not a non-Ewing’s cell line, U2OS, as measure by an MTT assay. (B) Using cell viability assays, inhibition of PARP with a drug olaparib combined with DNA damage induced by SN-38, which is currently in clinical trials, enhances cell death at 72 hrs but not at 12 hrs. (C) Inhibiting FET family co-factors involved in transcription (MR03) dramatically enhances cell death due to DNA damage for Ewing’s sarcoma cells after only 12 hours of treatment.

Tucson Web Design - CS Design Studios
Copyright © 2020 - theschwartzlab.com
Privacy Policy